- Latest news▼
-
15:32, April 26 ESCMID: New method of purifying the air with ultraviolet light could protect world from new pandemic

-
08:43, April 26 Enzymes that convert different blood groups into first group are discovered

-
19:41, April 25 Children’s Hospital Los Angeles and International Center of Professional Development Allergy/Immunology Conference

-
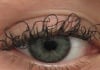
17:31, April 25 JAMA: patient grew long, curly eyelashes because of chemotherapy
-
11:08, April 25 Mpox epidemic declared in Republic of the Congo

-
08:31, April 25 OU: quitting smoking 4 times more likely to cure laryngeal cancer

-
01:20, April 25 Paralyzed man in China writes hieroglyphs using neural implants placed in his brain

-
15:11, April 24 Zombie deer disease possibly linked to hunters’ deaths

-
12:27, April 23 Appetite: Scientists found out the secret to the appeal of large portions of fast food

-
10:33, April 23 Scientists test new approach to fighting viruses

-
08:38, April 23 Ketamine may help with postpartum depression

-
22:12, April 22 Unhealthy amount of sugar found in baby food products of a well-known brand

-
19:41, April 22 Air pollution puts health of more than 1.6 billion workers globally at risk

-
17:25, April 22 Scientists found baked goods and lack of sleep to be more dangerous than alcohol

-
16:02, April 22 342 cases of measles recorded in Armenia so far in 2024
All materials
De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis

We previously established the contribution of de novo damaging sequence variants to Tourette disorder (TD) through whole-exome sequencing of 511 trios. Here, we sequence an additional 291 TD trios and analyze the combined set of 802 trios. We observe an overrepresentation of de novo damaging variants in simplex, but not multiplex, families; we identify a high-confidence TD risk gene, CELSR3 (cadherin EGF LAG seven-pass G-type receptor 3); we find that the genes mutated in TD patients are enriched for those related to cell polarity, suggesting a common pathway underlying pathobiology; and we confirm a statistically significant excess of de novo copy number variants in TD. Finally, we identify significant overlap of de novo sequence variants between TD and obsessive-compulsive disorder and de novo copy number variants between TD and autism spectrum disorder, consistent with shared genetic risk.
To follow up our phase 1 study (Willsey et al., 2017), we conducted WES on 291 new “phase 2” TD trios (802 total trios across phase 1 and 2). We also analyzed 582 new phase 2 control trios from the Simons Simplex Collection (SSC) (1,184 total control trios across phase 1 and 2). After quality control, we trimmed to 777 TD trios and 1,153 SSC trios for de novo sequence variant calling
Cohort characteristics as well as sequencing metrics are summarized per cohort and by phase. 95% confidence intervals are displayed as ±, where relevant.
We leveraged GATK to conduct alignment, quality control, and variant calling (DePristo et al., 2011, McKenna et al., 2010, Van der Auwera et al., 2013). We conducted joint genotyping across the entire set of phase 1 and phase 2 TD trios, as well as the entire set of control trios, in order to reduce batch effects. We further modified our previous de novo calling pipeline (Willsey et al., 2017) to utilize the GATK genotype refinement workflow. We defined likely gene disrupting (LGD) variants as insertion of a premature stop codon, disruption of a canonical splice site, or a frameshift insertion or deletion, and probably damaging missense 3 (Mis 3) variants include missense variants with a PolyPhen2 (HDIV) score ≥ 0.957 (Adzhubei et al., 2010, Adzhubei et al., 2013). We refer to the set of LGD and Mis3 variants as “damaging”.
We detected 309 de novo coding variants from phase 2 samples (1.09 variants per sample). Applying the new pipeline to the phase 1 samples, we detected a total of 466 de novo coding variants (0.94 variants per sample). The number of de novo variants per individual followed a Poisson distribution , and our new pipeline achieved a 95.9% validation rate across phase 1 and 2 TD samples. See STAR Methods for more details. We did not validate the de novo variants in control samples, and therefore, we conducted all burden analyses using all de novo variants identified in TD and control trios. However, for gene discovery, we considered validated de novo variants only. WES coverage varied across cohorts and phases because of the different capture arrays and sequencing protocols used and was positively correlated with the number of de novo variants observed per individual (STAR Methods). To account for these differences, we compared mutation rates, instead of the number of de novo variants observed per individual, to normalize for the number of bases with sufficient joint coverage for de novo calling (Willsey et al., 2017). To further reduce biases, we estimated mutation rates within a high-confidence region with high joint coverage across all cohorts (consensus region; Table 1; STAR Methods). We then compared the rate between TD probands and SSC siblings with a one-sided rate ratio test, as previously described (Willsey et al., 2017). We also confirmed that the overall rate of coding de novo sequence variants does not differ between phase 1 and phase 2 TD trios (rate ratio [RR] 1.03; p = 0.81; two-sided rate ratio test).
We excluded any de novo variants located outside of the consensus regions and then calculated the mutation rate per base pair and 95% CI using t test in R. See also Figures S4 and S5. Comorbid, probands with TD and ADHD/OCD; damaging, LGD + Mis3; in frame, indel causing in-frame deletion or insertion (loss or gain of amino acids); intolerant LGD, de novo LGD variants occurring in genes with pLI greater than 0.9; intolerant Mis, de novo missense variants occurring in genes with missense Z score greater than 3.891; intolerant Nonsyn, intolerant Mis + intolerant LGD; LGD, likely gene disrupting (insertion of premature stop codon, disruption of canonical splice site, and insertion-deletion frameshift); LGD FS, insertion-deletion variant causing frameshift; LGD SNV, point mutation causing insertion of premature stop codon and disruption of canonical splice site; Nonsyn, nonsynonymous; simplex, parents unaffected with TD; Syn, synonymous; unknown, phenotypic data unavailable for parents.
Follow NEWS.am Medicine on Facebook and Twitter

- Video



- Event calendar
- Children’s Hospital Los Angeles and International Center of Professional Development Allergy/Immunology Conference
- First Armenian-German Conference entitled “Heart Failure Spring School”
- Allogeneic bone marrow transplant in case of hematological malignancy performed in Armenia for first time
All materials
- Archive
- Most read
month
week
day
- Scientists found baked goods and lack of sleep to be more dangerous than alcohol 1026
- Next pandemic likely to be triggered by flu - scientists 994
- 342 cases of measles recorded in Armenia so far in 2024 951
- Scientists develop new method to safely stimulate immune cells to fight cancer 810
- Cognitively stimulating jobs in midlife could lower dementia risk in old age, study finds 808
- Blood test can determine who is at risk of developing multiple sclerosis - scientists 807
- Air pollution puts health of more than 1.6 billion workers globally at risk 761
- Unhealthy amount of sugar found in baby food products of a well-known brand 757
- Ketamine may help with postpartum depression 756
- Appetite: Scientists found out the secret to the appeal of large portions of fast food 749
- BrainStimulation: electrical brain stimulation alleviates anxiety and depression in the elderly 741
- Scientists test new approach to fighting viruses 732
- Zombie deer disease possibly linked to hunters’ deaths 664
- Mpox epidemic declared in Republic of the Congo 461
- Paralyzed man in China writes hieroglyphs using neural implants placed in his brain 418
- Find us on Facebook
- Poll