- Latest news▼
-
19:41, April 25 Children’s Hospital Los Angeles and International Center of Professional Development Allergy/Immunology Conference

-
17:31, April 25 JAMA: patient grew long, curly eyelashes because of chemotherapy
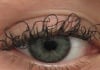
-
11:08, April 25 Mpox epidemic declared in Republic of the Congo

-
08:31, April 25 OU: quitting smoking 4 times more likely to cure laryngeal cancer

-
01:20, April 25 Paralyzed man in China writes hieroglyphs using neural implants placed in his brain

-
15:11, April 24 Zombie deer disease possibly linked to hunters’ deaths

-
12:27, April 23 Appetite: Scientists found out the secret to the appeal of large portions of fast food

-
10:33, April 23 Scientists test new approach to fighting viruses

-
08:38, April 23 Ketamine may help with postpartum depression

-
22:12, April 22 Unhealthy amount of sugar found in baby food products of a well-known brand

-
19:41, April 22 Air pollution puts health of more than 1.6 billion workers globally at risk

-
17:25, April 22 Scientists found baked goods and lack of sleep to be more dangerous than alcohol

-
16:02, April 22 342 cases of measles recorded in Armenia so far in 2024
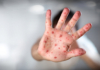
-
15:29, April 22 BrainStimulation: electrical brain stimulation alleviates anxiety and depression in the elderly

-
08:27, April 22 Cognitively stimulating jobs in midlife could lower dementia risk in old age, study finds

All materials
Treatment of a metabolic liver disease by in vivo genome base editing in adult mice
CRISPR–Cas-based genome editing holds great promise for targeting genetic disorders, including inborn errors of hepatocyte metabolism.
Precise correction of disease-causing mutations in adult tissues in vivo, however, is challenging. It requires repair of Cas9-induced double-stranded DNA (dsDNA) breaks by homology-directed mechanisms, which are highly inefficient in nondividing cells. Here we corrected the disease phenotype of adult phenylalanine hydroxylase (Pah)enu2 mice, a model for the human autosomal recessive liver disease phenylketonuria (PKU)1, using recently developed CRISPR–Cas-associated base editors.
These systems enable conversion of C∙G to T∙A base pairs and vice versa, independent of dsDNA break formation and homology-directed repair (HDR). We engineered and validated an intein-split base editor, which allows splitting of the fusion protein into two parts, thereby circumventing the limited cargo capacity of adeno-associated virus (AAV) vectors. Intravenous injection of AAV-base editor systems resulted in Pahenu2 gene correction rates that restored physiological blood phenylalanine (L-Phe) levels below 120 µmol/l. We observed mRNA correction rates up to 63%, restoration of phenylalanine hydroxylase (PAH) enzyme activity, and reversion of the light fur phenotype in Pahenu2 mice.
Our findings suggest that targeting genetic diseases in vivo using AAV-mediated delivery of base-editing agents is feasible, demonstrating potential for therapeutic application.
Follow NEWS.am Medicine on Facebook and Twitter

- Video



- Event calendar
- Children’s Hospital Los Angeles and International Center of Professional Development Allergy/Immunology Conference
- First Armenian-German Conference entitled “Heart Failure Spring School”
- Allogeneic bone marrow transplant in case of hematological malignancy performed in Armenia for first time
All materials
- Archive
- Most read
month
week
day
- JAMA Oncology: Urine test can help rule out high-grade prostate cancer with almost 100% accuracy, study shows 1312
- Scientists grow human mini-lungs in lab 1178
- Next pandemic likely to be triggered by flu - scientists 963
- Scientists found baked goods and lack of sleep to be more dangerous than alcohol 940
- 342 cases of measles recorded in Armenia so far in 2024 896
- Scientists develop new method to safely stimulate immune cells to fight cancer 786
- Cognitively stimulating jobs in midlife could lower dementia risk in old age, study finds 784
- Blood test can determine who is at risk of developing multiple sclerosis - scientists 782
- BrainStimulation: electrical brain stimulation alleviates anxiety and depression in the elderly 722
- Ketamine may help with postpartum depression 676
- Unhealthy amount of sugar found in baby food products of a well-known brand 675
- Appetite: Scientists found out the secret to the appeal of large portions of fast food 666
- Air pollution puts health of more than 1.6 billion workers globally at risk 666
- Scientists test new approach to fighting viruses 653
- Zombie deer disease possibly linked to hunters’ deaths 567
- Find us on Facebook
- Poll